|
|
|
|
Primer on Illumina Data Reduction
|
|
|
|
 | This document summarizes how I analyze raw Illumina data on an Apple Mac Pro. I often refer often to the analysis of strain TT26511. The page you are reading was exported as HTML from my electronic notebook, created using the application Notebook ($50, http://www.circusponies.com/). Notebook is an excellent way to keep track of the many steps and copious data generated during data reduction.
|
|
|
|
|
| Aside from Microsoft Excel, the remaining software described in this primer is free and in the public domain (or a component of the OS X operating system).
|
|
|
|
|
 | When you installed the latest version of OS X on your computer, you probably did the easy install, in which case, you should do the following:
|
|
|
|
|
| 1. Insert the Mac OS X installation disc
|
|
|
|
|
| 2. Double-click the Optional Installs package
|
|
|
|
|
| 3. Follow the onscreen instructions to install "XCode Tools" and "X11". This will will make your life easier.
|
|
|
|
|
| Illumina data reduction requires commands run in a terminal window provided by the Terminal application (/Applications/Utilities). You will need to be familiar with the following when using this program. All are intrinsic parts of the underlying Unix foundation of OS X.
|
|
|
|
|
| You can download a stand-alone version of Emacs from http://emacsformacosx.com. This is a more flexible and efficient version of the program than the one provided by the OS.
|
|
|
|
|
| Terminal commands in this primer are usually prefixed with "$ " to indicate the standard terminal prompt. This symbol should not be copied when trying out the command.
|
|
|
|
|
| Some of my tools are written in Python. No knowledge of this language is required to run them, but it is important that the version number be 2.7 or higher and less than 3.0. The latter is actually a different language and is incompatible with my setup.
|
|
|
|
|
| You can test your version of Python by running "python --version" in Terminal. If it is less than 2.7, you can upgrade by using MacPorts. This is much easier than trying to install it from the official Python site, as all the prerequisites required under OS X are taken care of automatically (hence, the "Mac" in MacPorts).
|
|
|
|
|
| I use the "maq" suite of DNA analysis programs to manipulate Illumina data. They were recommended highly by Yong Lu in Fritz Roth's lab and have proven invaluable in my work. Maq can be downloaded here. To learn more about it, consult the manual.
|
|
|
|
|
| Unix variables can be created which will allow easy access to directories, databases, etc.
|
|
|
|
|
| The following illustrates the bash code for setting variables pointing to data directories in the analysis of TT26511.
|
|
|
|
|
| $ export tt26511=/Users/kofoid/projects/inverted_duplications/Solexa/3_5_12/tt26511
|
|
|
|
|
| $ export dtt26511=/Users/kofoid/projects/inverted_duplications/Solexa/dData/d3_5_12/dtt26511
|
|
|
|
|
| To see the value of a Unix variable, prefix the variable name with "$". In the following examples, "echo" is the bash word for "print":
|
|
|
|
|
| $ foo=123
$ echo foo
-bash: foo: No such file or directory
$ echo $foo
123
|
|
|
|
|
| The Unix command for "connect to directory" is "cd". To go to folder /Users/kofoid/projects/inverted_duplications/Solexa/3_5_12/tt26511, do the following:
|
|
|
|
|
| To be useful, command lines creating variables must be stored in a bash startup file, which is loaded whenever you begin a session with Terminal. I use a file called ".bash_temp" in my home directory (Unix abbreviation "~"), and insert the following line in the startup file "~/.bash_profile" to call it:
|
|
|
|
|
| . ~/.bash_temp # there is a space between the dot and tilde; it's important!
|
|
|
|
|
| Here are a few lines from my .bash_temp file:
|
|
|
|
|
| # Temporary shortcuts:
export sq419=~/projects/inverted_duplications/Solexa/10_27_10/sq419
export dsq419=~/projects/inverted_duplications/Solexa/dData/d10_27_10/dsq419
export tt26511=/Users/kofoid/projects/inverted_duplications/Solexa/3_5_12/tt26511
export dtt26511=/Users/kofoid/projects/inverted_duplications/Solexa/dData/d3_5_12/dtt26511
# Temporary blast db's:
export sq419db=$dsq419/sq419
export tt26511db=$dtt26511/tt26511db
...etc
|
|
|
|
|
| These steps convert the data to forms which allow easy construction of read-depth profiles, SNP discovery, description of rearrangements, etc.
|
|
|
|
|
| The raw data you receive can be in any one of a number of formats, depending on the kind of preprocessing done by your sequencing facility. My sample page assumes a file in zipped Illumina fastq format, segmented into multiple files. The following steps uncompress, merge and convert the data into the Sanger fastq format used by maq:
|
|
|
|
|
| $ mv TT26511_GAGTGG_L007_R1_001 TT26511_GAGTGG_L007_R1.fastq
|
|
|
|
|
| $ cat *_R1_* >> TT26511_GAGTGG_L007_R1.fastq
|
|
|
|
|
| $ mv TT26511_GAGTGG_L007_R2_001 TT26511_GAGTGG_L007_R2.fastq
|
|
|
|
|
| $ cat *_R2_* >> TT26511_GAGTGG_L007_R2.fastq
|
|
|
|
|
| $ maq sol2sanger TT26511_GAGTGG_L007_R1.fastq $dtt26511/foo_r1.fastq
|
|
|
|
|
| $ maq sol2sanger TT26511_GAGTGG_L007_R2.fastq $dtt26511/foo_r2.fastq
|
|
|
|
|
| At this point, you should check the data for suffices "#0/1" or "#0/2" at the ends of descriptors for left or right reads, respectively. Many facilities remove these during preprocessing.
|
|
|
|
|
| If they are not present, the following code will restore them. You will have to replace "@DJB" with whatever begins your particular data descriptor lines:
|
|
|
|
|
| $ sed 's/\(@DJB.*\)/\1#0\/1/' foo_r1.fastq > tt26511_r1.fastq
|
|
|
|
|
| $ sed 's/\(@DJB.*\)/\1#0\/2/' foo_r2.fastq > tt26511_r2.fastq
|
|
|
|
|
| Otherwise, you can simply rename the files appropriately:
|
|
|
|
|
| $ mv foo_r1.fastq tt26511_r1.fastq
|
|
|
|
|
| $ mv foo_r2.fastq tt26511_r2.fastq
|
|
|
|
|
| Convert fastq to fasta, which will be used to build a Blast database (and can also be used directly as a FastA database). Again, you will have to substitute the correct descriptor prefix for "@DJB775P1":
|
|
|
|
|
| $ awk 'BEGIN { RS="@DJB775P1"; FS="\n" } ; { print ">DJB7751P1"$1"\n"$2 }' tt26511_r1.fastq > tt26511_r1.fasta
|
|
|
|
|
| $ awk 'BEGIN { RS="@DJB775P1"; FS="\n" } ; { print ">DJB7751P1"$1"\n"$2 }' tt26511_r2.fastq > tt26511_r2.fasta
|
|
|
|
|
| Convert fastq to the binary format, "bfq", used by many maq programs:
|
|
|
|
|
| $ maq fastq2bfq tt26511_r1.fastq tt26511_r1.bfq
|
|
|
|
|
| $ maq fastq2bfq tt26511_r2.fastq tt26511_r2.bfq
|
|
|
|
|
| You can search your data for specific targets using a local copy of the NCBI Blast program, which can be downloaded here. This also installs a variety of tools, including "makeblastdb" which can be used to create a Blast database from fasta-formatted data:
|
|
|
|
|
| $ cp tt26511_r1.fasta foo.fa
|
|
|
|
|
| $ cat tt26511_r2.fasta >> foo.fa
|
|
|
|
|
| $ makeblastdb -in=foo.fa -max_file_sz=80000000 -title=tt26511db -dbtype=nucl -out=tt26511db
|
|
|
|
|
| $ export tt26511db=$dtt26511/tt26511db
|
|
|
|
|
| Note that I also created a Unix variable which points to the location of the database, and stored the line creating it in my .bash_temp file (see above).
|
|
|
|
|
| I have written a blast front end, "bl", which allows easy command line blast searches. The code is here.
|
|
|
|
|
| Typical Blast search using the above variable for the database:
|
|
|
|
|
| $ bl foo.fasta $tt26511db
|
|
|
|
|
| Output will go to the file "foo.blast" in the current working directory (i.e., wherever you are at the moment in Terminal).
|
|
|
|
|
 | A comment about file sizes
|
|
|
|
|
| Everything derived from the raw data files is really big! You should have a backup strategy for the base fastq files created after expanding the compressed raw files. If you use Time Machine (TM), then leave the fastq files in the current directory. Because you will never change them, TM will back them up once and never again, which is good. You should also copy them to a portable hard disk which you can keep off site (or at least in another room). Your sequencing facility will probably keep the original files for a few weeks to months, but don't rely on them to store your data forever.
|
|
|
|
|
| All the derived files (Sanger fastq, fasta, bfq, blast, etc.) can be recreated if lost, and would only take up unnecessary space in the TM archive. Notice that when I did the sol2sanger step, the Sanger fastq files were placed in a different directory, $dtt26511, after which I moved to that directory ("cd $dtt26511"). All other big files were also stored there. I have instructed TM to not backup anything in folders beginning with "d", which indicates "don't archive".
|
|
|
|
|
| Here, we finally do the good stuff.
|
|
|
|
|
| Remember, we're in the "big file" directory. First, I create a subdirectory for chromosomal stuff, "dsty", and move into it:
|
|
|
|
|
| Now I run a workhorse script, "sol", which does all the rest of the work and spits out a plot, a histogram file readable by Excel, and some searchable "view" files. The code is here. This particular script calls several maq programs, and ultimately generates a plot by calling a script "rdplot", the code for which is here.
|
|
|
|
|
| rdplot calls several gnuplot routines, which means you will need to have gnuplot on your system. You can check for it by simply typing "gnuplot --version" in Terminal. If you get something like "gnuplot 4.6 patchlevel 1" as a response, you're set. Otherwise, you can install gnuplot from MacPorts, as suggested above for Python.
|
|
|
|
|
| To see the plot, you will also have to have the utility program, "AquaTerm". It may already exist in /Applications/Utilities, but you will probably want to install the newest version using MacPorts.
|
|
|
|
|
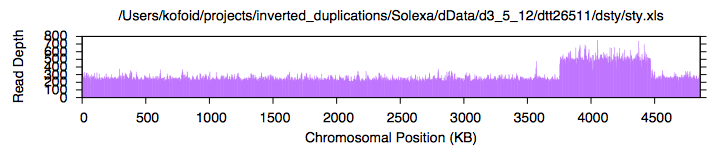
| If "sol" runs successfully, you will see some amusing lines of information scroll up in the terminal window, and eventually a plot will appear in an AquaTerm graphic window which looks something like this:
|
|
|
|
|
| In general, I have tried to avoid using any proprietary software. However, in the sample page for TT26511, you will notice some statistics regarding read depth, such as "overall read depth", "standard deviation of read depth", "amplification ratio", etc. My quick-and-dirty method for calculating these uses Excel. Eventually, I'll write a script to do this, but don't hold your breath.
|
|
|
|
|
| "sol" produces a file with extension ".xls" which contains the binned data necessary to generate the above plot, It can also be used to calculate statistics in Excel.
|
|
|
|
 |
|
| Here is a typical Excel file used for this purpose:

|
|
|
|
|
| Open the ".xls" file in Excel and copy the data in column A.
|
|
|
|
|
| Use "paste special/values" to insert the data into column B of the ".xlsx" file, overwritting any previous data. You may have to adjust the ranges of the other columns (A, C & D) to correspond to the new data.
|
|
|
|
|
| You will immediately notice a plot at the top of the worksheet, essentially identical to the one created earlier. If you needed to change data ranges to correspond to the new data, you will also have to change the ranges over which this plot is made.
|
|
|
|
|
| Make sure that the "Bin Size" correspondes to the same-named parameter given to "sol".
|
|
|
|
|
| Under the plot are several lines of statistics. Examine the formulae underlying them, and adjust the data ranges to correspond to the amplification features in the plot. More exact estimates of edges can be found by scrolling down through column B and finding the positions where read-depth suddenly changes.
|
|
|
|
|
|
| Positional normalization is a method for removing much of the noise from a read-depth plot. To use this technique, you must also have sequenced an unselected parental control strain along with the test strains. Here is an Excel file with normalization:

|
|
|
|
|
| To use it, do the same manipulations as for a normal Excel session. Then, use "paste special/values" to overwrite the data in column N with the data from column A of the control ".xls" file.
|
|
|
|
|
| The lower plot will automagically convert to a smoothed version of the upper.
|
|
|
|
|
|
| You will notice in the TT26511 sample page that I also run analyses for anomalous features using Yong Lu's "find-anamo" perl script. I won't go into details here, but the tool can be created from the following file ("find_unmapped_pair.ml"):

|
|
|
|
|
| Here are Yong's instructions for installing it, paraphrased slightly:
|
|
|
|
|
| 1. Using Terminal, move to the directory containing the file (using "cd").
|
|
|
|
|
| 2. $ chmod ugo+rwx find_unmapped_pair.ml
|
|
|
|
|
| This changes the permissions of the file, essentially turning it into a runnable script.
|
|
|
|
|
| If Terminal balks at this command, prefix it with "sudo ", which turns you momentarily into superuser. You will have to give your Mac password to complete the command.
|
|
|
|
|
| 3. A file with extension ".exe" will appear in the directory. Change its entire name, including the extension, to "find-anamo" and move it to a useful final directory.
|
|
|
|
|
| I did "sudo mv find-anamo /usr/bin/maq", but you may have a better place.
|
|
|
|
|
| Wherever you put it, it should be on the path defined by your $PATH variable.
|
|
|
|
|
| If this makes no sense to you, read your Unix book or ask your IT person.
|
|
|
|
|
| This is a Swiss Army knife for locating reads in ".view" files subject to various criteria. The Python code for it can be found here. It has 4 modes of use:
|
|
|
|
|
| 1. Locate a range of IDs. The input file must be sorted by ID first:
|
|
|
|
|
| $ getfv -i1 <ID 1> [-i2 <ID 2>] [-p <period>] [-v] <infile> > <outfile>
|
|
|
|
|
| 2. Locate a range of addresses (positions in the reference sequence). The input file must be sorted by position first,although this is the default when view files are created:
|
|
|
|
|
| $ getfv -p1 <pos. 1> [-p2 <pos. 2>] [-p <period>] [-v] <infile> > <outfile>
|
|
|
|
|
| 3. Locate a range of lines. This is rarely useful:
|
|
|
|
|
| $ getfv -l1 <line 1> [-l2 <line 2>] [-p <period>] [-v] <infile> > <outfile>
|
|
|
|
|
| 4. Locate IDs listed in a text file. The input must be sorted by ID first:
|
|
|
|
|
| $ getfv -f <ID file> [-p <period>] [-v] <infile> > <outfile>
|
|
|
|
|
| Output will be in multi-fasta format unless the flag "-V" is present on the command line, in which case the view format is preserved.
|
|
|
|
|
| Yes, you are right -- this should have been written as four separate tools! I will get around to it eventually.
|
|
|
|
|
| This will find overlapping contigs in a group of reads.
|
|
|
|
|
| Download cap3 here, and follow the directions in the README file.
|
|
|
|
|
| $ cap3 <fasta reads file>
|
|
|
|
|
| For weak contigs, I often use:
|
|
|
|
|
| $ cap3 <fasta reads file> -d 400 -e 40 -m 3 -o 30 -p 75 -s 750
|
|
|
|
|
| several output files are created, but the important one ends ".contigs"
|
|
|
|
|
| FastA is a very sensitive and accurate program which seeks matches between a target sequence and records in a database. It is much slower than Blast.
|
|
|
|
|
| However, it is quite useful under certain conditions:
|
|
|
|
|
| 1. The number of database records is not too large.
|
|
|
|
|
| 2. It's not worth the effort to create a Blast database and use Blast.
|
|
|
|
|
| 3. The records are stored as a collection of fasta-formatted files.
|
|
|
|
|
| I have written a front end to FastA, "fa", which simplifies using the program from the command line. The code is here, and "fa -h" will provide help on using it. More extensive help can be got by running "fa -help".
|
|
|
|
|
| There are several ways to indicate the databases being searched:
|
|
|
|
|
| Multifasta: Any file containing many fasta-formatted records can be used as a fasta database. Here are the first few lines of my primer database, "tp.fa":
|
|
|
|
|
| >gnl|pr|1 1061ad-8;TP1
AGGGTTGCCGGAAAATTTCCAT
>gnl|pr|10 a1w;TP10
GACCATCGCCAGCCA
>gnl|pr|100 gent2;TP100
GGGGATCCGCATAAAGCGGGTGATGGGA
>gnl|pr|1000 deor2;TP1000
AAGGCTGGGTACGTGACTAA
>gnl|pr|1001 hspt1;TP1001
TAACCACTTAAATTCGTGACTAAAAAAAGGCTTTTATAGACACCAAACACCCCCCAAAAC
...etc
|
|
|
|
|
| Blast: fa can search a blast database in the following fashion:
|
|
|
|
|
| Create a file containing the unique prefices of all the files in the Blast database followed by " 12". As an example, the first few lines for such a file drawn from the TT26511 data would be:
|
|
|
|
|
| tt26511db.00 12
tt26511db.01 12
tt26511db.02 12
tt26511db.03 12
tt26511db.04 12
tt26511db.05 12
tt26511db.06 12
tt26511db.07 12
tt26511db.08 12
tt26511db.09 12
...etc
|
|
|
|
|
| The " 12" field tells FastA that the data is part of a Blast database.
|
|
|
|
|
| Use this file as follows (assuming its name is "tt26511.fa"):
|
|
|
|
|
| $ fa foo.fasta @tt26511.fa
|
|
|
|
|
| The "@" in front of the file name is important; it tells the program to expect a list of file names rather than data.
|
|
|
|
|
| Dynamic: fa can directly search a directory containing many fasta-formatted files, as shown in this example:
|
|
|
|
|
| $ fa foo.fasta $prm/tp*fasta
|
|
|
|
|
| The target is "foo.fasta" and a temporary database is constructed dynamically from all the files in directory $prm which match the pattern "tp*fasta", where the asterisk is a wildcard meaning "any text". This is, in fact, all of the primers in the Rothlab collection, stored according to TP number.
|
|
|
|
|
| Working remotely with X11
|
|
|
|
|
| This discussion assumes that your bioinformatic software is running on a server under OS X, always kept on, and configured for file sharing as follows:
|
|
|
|
|
| Open Apple/System Preferences/Sharing and check the "File Sharing" box
|
|
|
|
|
| Click on Options and check the "Share files and folders using AFP", and dismiss by clicking "Done"
|
|
|
|
|
| Close the Preferences window
|
|
|
|
|
| From the client machine (an OS X computer remote from the server), link to the server using "Finder/Go/Connect to Server..."
|
|
|
|
|
| Fill in the server IP address and click "Connect".
|
|
|
|
|
| Mount the server itself from the list presented and click "OK".
|
|
|
|
|
| Drag-and-drop and other file manipulations are now enabled between client and server.
|
|
|
|
|
| Open the Terminal program and connect to the server as follows:
|
|
|
|
|
| $ ssh -Y <login name>@<server IP address or domain name>
|
|
|
|
|
| <login name> is the login name use when sitting at the server itself and restarting it.
|
|
|
|
|
| <server IP address> can be found at the server in "Apple/System Preferences/Network".
|
|
|
|
|
| <domain name> can be found at the server in "Apple/System Preferences/Sharing/File Sharing" in the text between the double slash and single slash under the "File Sharing: On" radio button.
|
|
|
|
|
| Enter the password (the same used to restart the server as a computer).
|
|
|
|
|
| The terminal window now belongs to the server and the work you do will be exactly the same as if you were at the server computer itself.
|
|
|
|
|
| Any files created (for instance, the postscript file containing the image of the read-depth profile) can be inspected in the remote server window previously opened.
|
|
|
|
|
| You will not be able to see the AquaTerm image of the read-depth profile, as AquaTerm is a native OS X application, and will not work across the X11 interface. However, if you double click the postscript file made during the run, an identical image will open in Preview on the client machine.
|
|
|
|
|
| Likewise, the stand-alone version of Emacs will work across the network connection. Instead, the terminal version of Emacs or the local stand-alone applicaiton must be used.
|
|
|
|
|
| Using Emacs at the terminal iis most easily done by opening two server sessions and dedicating one to Emacs alone.
|
|
|
|
|
| Alternatively, a remote file can be opened in the client Emacs application. This can be sluggish as the file being edited moves back and forth between the two computers whenever it is saved or opened. The larger the file, the greater the problem.
|
|
|
|
|
| Excel work should be done on copies of files dragged from the server window into a client window. When Excel works on a remote file directly, performance degrades rapidly because of autosave, caching, and other problems.
|
|
|
|
|
| Be sure to copy the file back to the server when done!
|
|
|
|
|
| On a couple of occasions, I have written master scripts which automate everything, from raw data to the final profile. Invariably, they screw up, often with loss of data. The lesson: Don't try to do too much in a single step! Similarly, try to convince your sequence facility to keep your raw data on their server as long as possible, just in case you have to fetch it again.
|
|
|
|
|
| Many of these procedures and tools are in flux and being improved. I'm happy to hear comments and suggestions, especially about improvements in my code.
|
|
|
|

|
 |
|